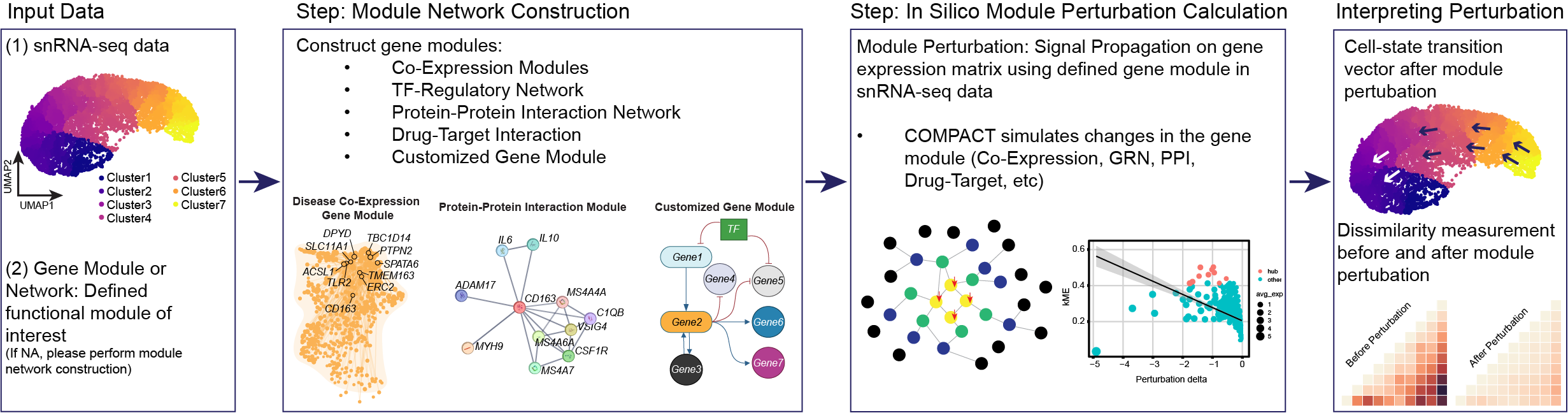
compact: co-expression module perturbation analysis


compact is a framework for performing CO-expression Module Perturbation Analysis in Cellular Transcriptomes. Building off of our previous method hdWGCNA, compact applies direct perturbations to co-expression network hub genes, and uses the network structure to propagate the perturbation signal to other linked genes in the network. This framework is highly flexible to perform knock-in, knock-down, or knock-out perturbations on different networks and sets of genes and in different cell lineages, allowing researchers to explore a wide range of strategies mimicking various experimental conditions and interventions.
compact is under active development, and is currently an alpha version. This means that many features of the final package are missing, and current features are subject to change throughout development. The first major release of compact will coincide with our forthcoming publication.
To get started, please install the package and then visit the co-expression module perturbation analysis tutorial.
Installation
Follow these steps to create an R conda environment environment for compact.
# Create a new conda environment, and activate it
conda create -n compact -c conda-forge -c bioconda r-base=4.4 mamba
conda activate compact
# Install Key packages via conda-forge
mamba install -c conda-forge -c bioconda \
r-seurat \
r-velocyto.r \
bioconductor-singlecellexperiment \
bioconductor-pcamethods \
r-hdf5r
# Install additional required packages
mamba install -c conda-forge \
r-devtools r-remotes r-tidyverse r-patchwork r-matrix r-rcpparmadillo r-ggpubr r-lme4 r-nloptr r-rstatix r-car r-pbkrtest r-ggrastrNext, install the required packages inside of R.
# install BiocManager
install.packages("BiocManager")
# install hdWGCNA
BiocManager::install(c("WGCNA", "UCell", "GenomicRanges", "GeneOverlap"))
devtools::install_github('smorabit/hdWGCNA', ref='dev')
# finally, install compact
devtools::install_github("velocyto-team/velocyto.R")
devtools::install_github('smorabit/compact')Install additional packages for Transcription Factor network analysis.
mamba install -c conda-forge -c bioconda bioconductor-tfbstools bioconductor-rtracklayer bioconductor-genomicranges bioconductor-motifmatchrWithin R:
BiocManager::install(c(
'EnsDb.Hsapiens.v86',
'BSgenome.Hsapiens.UCSC.hg38',
'JASPAR2020',
'JASPAR2024'
))